
Как сделать секвенирование по сэнгеру
Добавил пользователь Владимир З. Обновлено: 04.10.2024
Секвенирование ДНК – это процесс определения последовательности нуклеотидов в ДНК молекула, каждый организм ДНК состоит из уникальной последовательности нуклеотидов. Определение последовательности может помочь ученым сравнить ДНК между организмами, что может помочь показать, как эти организмы связаны.
Обзор секвенирования ДНК
Это означает, что, упорядочив отрезок ДНК, можно будет узнать порядок, в котором четыре нуклеотид основы – аденин, гуанин, цитозин и тимин – происходят в этом нуклеиновая кислота молекулы.
Необходимость секвенирования ДНК впервые стала очевидной из теории Фрэнсиса Крика о том, что последовательность нуклеотидов в молекуле ДНК непосредственно влияет на аминокислотные последовательности белков. В то время считалось, что полностью секвенированный геном приведет к квантовому скачку в понимании биохимия клеток и организмов.
Пример секвенирования ДНК
Хотя секвенирование ДНК раньше занимало годы, теперь это можно сделать за часы. Далее, первая полная последовательность человеческой ДНК заняла около 3 миллиардов долларов. Теперь некоторые компании упорядочат весь ваш геном менее чем за 1000 долларов. Самые продвинутые тесты будут анализировать каждый нуклеотид в вашем геноме. Тем не менее, многие компании сейчас предлагают однонуклеотидный полиморфизм тесты.
Эти тесты фокусируются на отдельных нуклеотидах в генах, которые могут обозначать определенные генетические варианты. Эти SNP, как они известны, связаны с определенными условиями и могут помочь предсказать, как ваши гены могут влиять на вашу жизнь. Некоторые SNP связаны с различными заболеваниями, в то время как другие связаны с вашим метаболизмом и тем, как ваш организм перерабатывает питательные вещества. Были найдены тысячи различных корреляций, и можно использовать секвенирование ДНК, чтобы выяснить, как ваш геном влияет на вашу жизнь.
Методы секвенирования ДНК
Существует два основных типа секвенирования ДНК. Более старый, классический метод обрыва цепи также называется методом Сэнгера. Новые методы, которые могут быстро обрабатывать большое количество молекул ДНК, в совокупности называются методами высокопроизводительного секвенирования (HTS) или методами секвенирования следующего поколения (NGS).
Секвенирование
Метод Сэнгера опирается на праймер, который связывается с молекулой денатурированной ДНК и инициирует синтез одноцепочечного полинуклеотида в присутствии фермента ДНК-полимеразы, используя денатурированную ДНК в качестве матрицы. В большинстве случаев фермент катализирует добавление нуклеотида. Ковалентная связь поэтому образует между 3 ‘атомом углерода дезоксирибоза молекула сахара в одном нуклеотиде и 5 ‘атом углерода в следующем. Это изображение ниже показывает, как формируется эта связь.

В самых ранних попытках использования метода Сэнгера молекулу ДНК сначала амплифицировали, используя меченый праймер, а затем разделяли на четыре пробирки, каждая из которых имела только один тип ddNTP. То есть каждая реакционная смесь будет иметь только один тип модифицированного нуклеотида, который может вызвать обрыв цепи. После того, как четыре реакции были завершены, смесь молекул ДНК, созданная путем обрыва цепи, подвергалась электрофорезу в полиакриламидном геле и разделялась в соответствии с их длиной.
Со временем этот метод был модифицирован таким образом, чтобы каждый ddNTP имел различную флуоресцентную метку. Праймер больше не был источником радиоактивной метки или флуоресцентной метки. Этот метод, также известный как секвенирование терминатора красителя, использовал четыре красителя с неперекрывающимися спектрами излучения, по одному для каждого ddNTP.


Высокая пропускная способность
Секвенирование Сэнгера продолжает быть полезным для определения последовательностей относительно длинных участков ДНК, особенно в небольших объемах. Однако, это может стать дорогим и трудоемким, когда большое количество молекул должно быть быстро секвенировано. Как ни странно, хотя традиционный метод терминатора красителя полезен, когда молекула ДНК длиннее, высокопроизводительные методы стали более широко использоваться, особенно когда необходимо секвенировать целые геномы.
Есть три основных изменения по сравнению с методом Сэнгера. Первой была разработка клетка система для клонирования фрагментов ДНК. Традиционно участок ДНК, который нужно было секвенировать, сначала клонировали в прокариот плазмида и усиливается в бактерии перед извлечением и очисткой. Технологии секвенирования с высокой пропускной способностью или секвенирования следующего поколения больше не полагались на эту трудоемкую и длительную процедуру.
Появление HTS значительно расширило приложения для геномики. Секвенирование ДНК стало неотъемлемой частью фундаментальной науки, трансляционных исследований, медицинской диагностики и криминалистики.
Использование секвенирования ДНК
С другой стороны, HTS позволяет использовать секвенирование ДНК для понимания однонуклеотидных полиморфизмов – среди самых распространенных типов генетическая изменчивость в пределах Население, Это становится важным в эволюционной биологии, а также в обнаружении мутировавших генов, которые могут привести к болезни. Например, вариации последовательности в образцах аденокарциномы легкого позволили обнаружить редкие мутации, связанные с заболеванием. Сайты связывания хроматина для специфических ядерных белков также могут быть точно идентифицированы с использованием этих методов.
В целом, секвенирование ДНК становится неотъемлемой частью многих различных приложений.
диагностика
Секвенирование генома особенно полезно для выявления причин редких генетических нарушений. В то время как более 7800 заболеваний связаны с паттерном менделевского наследования, менее 4000 из этих болезней были окончательно связаны с конкретным геном или мутация, Ранний анализ экзон -геном или экзом, состоящий из всех экспрессируемых генов организма, показал многообещающую роль в выявлении причинных аллелей для многих наследственных заболеваний. В одном конкретном случае секвенирование генома ребенка, страдающего тяжелой формой воспалительного заболевания кишечника, связало заболевание с мутацией в гене, связанном с воспалением – XIAP. В то время как у пациента первоначально были множественные симптомы, указывающие на иммунодефицит, по результатам секвенирования ДНК была рекомендована пересадка костного мозга. Ребенок впоследствии излечился от недуга.
Кроме того, HTS был важным игроком в развитии лучшего понимания опухолей и раковых заболеваний. Понимание генетической основы опухоли или рака позволяет врачам иметь в своем комплекте дополнительный инструмент для принятия диагностических решений. Атлас генома рака и Международный консорциум по геному рака секвенировали большое количество опухолей и продемонстрировали, что эти разрастания могут значительно различаться с точки зрения их мутационного ландшафта. Это также позволило лучше понять, какие варианты лечения являются идеальными для каждого пациента. Например, секвенирование генома рака молочной железы идентифицировало два гена – BRCA1 и BRCA2 – чьи патогенные варианты оказывают огромное влияние на вероятность развития рака молочной железы. Люди с некоторыми патогенными аллелями даже предпочитают проводить профилактические операции, такие как двойная мастэктомия.
Молекулярная биология
Секвенирование ДНК в настоящее время является неотъемлемой частью большинства биологических лабораторий. Он используется для проверки результатов упражнений на клонирование, чтобы понять влияние определенных генов. HTS-технологии используются для изучения вариаций генетического состава плазмид, бактерий, дрожжей, нематод или даже млекопитающих, используемых в лабораторных экспериментах. Например, клеточная линия, полученная из рака молочной железы ткань называется HeLa, используется во многих лабораториях по всему миру и ранее считался надежной клеточной линией, представляющей ткани молочной железы человека. Недавние результаты секвенирования продемонстрировали большие различия в геноме клеток HeLa из разных источников, тем самым уменьшая их полезность в клеточной и молекулярной биологии.
Секвенирование ДНК дает представление о регуляторных элементах в геноме каждой клетки и об изменениях их активности у разных типов клеток и индивидуумов. Например, определенный ген может быть постоянно выключен в некоторых тканях, в то время как конститутивно экспрессируется в других. Точно так же люди с восприимчивостью к определенному заболеванию могут регулировать ген иначе, чем те, кто обладает иммунитетом. Эти различия в регуляторных областях ДНК могут быть продемонстрированы с помощью секвенирования и могут дать представление о основе для фенотип.
Последние достижения даже позволили отдельным лабораториям изучать структурные изменения в геноме человека – мероприятии, которое потребовало глобального сотрудничества два десятилетия назад.
Криминалистика
Возможность использовать низкие концентрации ДНК для получения надежных результатов секвенирования была чрезвычайно полезна для судебно-медицинских экспертов. В частности, потенциал для последовательности каждой ДНК в образце является привлекательным, особенно потому, что место преступления часто содержит генетический материал от нескольких людей. HTS медленно внедряется во многих криминалистических лабораториях для идентификации человека. Кроме того, последние достижения позволяют судмедэксперту упорядочить экзом человека после смерти, особенно для определения причины смерти. Например, смерть от отравления покажет изменения экзома в пораженных органах. С другой стороны, секвенирование ДНК может также определить, что умерший имел ранее существовавшую генетическую болезнь или предрасположенность. Проблемы в этой области включают разработку чрезвычайно надежного программного обеспечения для анализа, тем более что результаты HTS не могут быть проверены вручную.
Секвенирование нуклеиновых кислот — это процесс определения последовательности нуклеотидов в цепочке ДНК.
Секвенирование нуклеиновых кислот — это процесс определения последовательности нуклеотидов в цепочке ДНК.
Основными задачами генетического анализа являются:
- обнаружение неизвестных ранее последовательностей нуклеиновых кислот, например, геномов нового типа;
- распознавание уникальных особенностей конкретного образца среди обобщённой последовательности (ресеквенирование);
- исследование генетического разнообразия, в том числе однонуклеотидных замен (SNP-типирование);
- определение эпигенетических модификаций, например, профиля метилирования;
- изучение профиля экспрессии отдельно взятых клеток секвенированием кДНК, полученной из тотальной мРНК клетки.
Сравнительные таблицы производительности секвенаторов
Капиллярные
Пиросеквенаторы
NGS-секвенаторы:
Генетические анализаторы различаются по ряду характеристик, среди которых:
- Длина прочтений.
- короткие;
- средние;
- длинные.
- Применяемая технология.
- технология по методу Сэнгера;
- пиросеквенирование;
- ионное полупроводниковое секвенирование;
- циклическое лигазное секвенирования;
- секвенирования на молекулярных кластерах с использованием флуоресцентно меченых нуклеотидов;
- одномолекулярное секвенирование.
- Число прочтений за запуск.
- Количество анализируемых одновременно маркеров.
- Время, необходимое для запуска.
- Стоимость самого прибора, одного запуска, анализа одной пробы.
Технологии секвенирования
На сегодняшний день существуют несколько основных методов считывания последовательности ДНК при помощи секвенаторов — классические и нового поколения.
Метод Сэнгера (ферментативный)
Синтез проводится в 4 разных пробирках с реакционной смесью, состоящей из праймера, стандартных дезоксинуклеотидов (dATP, dGTP, dCTP, dTTP), а также малого количества одного из четырёх радиоактивно меченых дезоксинуклеотидов (дидезоксинуклеотидов). Готовятся четыре раствора с каждым ddNTP по отдельности. У дидезоксирибонуклеотидов (ddATP, ddGTP, ddCTP, or ddTTP) отсутствует 3'-гидроксильная группа, поэтому после их включения в цепь дальнейший синтез обрывается.
Таким образом, в каждой пробирке образуется набор фрагментов ДНК разной длины, которые заканчиваются одним и тем же нуклеотидом (в соответствии с добавленным дидезоксинуклеотидом). После завершения реакции содержимое пробирок разделяют электрофорезом в полиакриламидном геле в денатурирующих условиях и проводят авторадиографию гелей.
На сегодняшний день секвенирование ДНК по Сэнгеру полностью автоматизировано. В настоящее время вместо радиоактивно меченных нуклеотидов используют дидезоксинуклеотиды с флуоресцентными метками с разными длинами волн испускания, благодаря этому реакцию можно проводить в одной пробирке.
Циклическое лигазное секвенирование
Технология циклического лигазного секвенирования (Sequencing by Oligonucleotide Ligation and Detection — SOLiD) разработана компанией Life Technologies и является коммерчески доступной с 2006 года. Этот метод можно разделить на следующие этапы: подготовка библиотек, ПЦР в эмульсии, насыщение бусин целевой ДНК, перенос бусин, секвенирование, расшифровка данных.
На первом этапе целевая ДНК разрезается на небольшие фрагменты, а затем к фрагментам пришиваются короткие нуклеотидные последовательности адаптеры А1 и А2. В итоге в библиотеке оказываются одноцепочечные нуклеотидные последовательности A1-(фрагмент ДНК)-A2.
После этого проводят ПЦР ДНК-фрагментов библиотеки в небольших каплях. Каждая капля содержит праймеры P1 и Р2, которые являются комплементарными по отношению к фрагментам А1 и А2. Каждая бусина несёт только один фрагмент праймера. В результате чего на каждой бусине оказывается по одной цепочке ДНК комплементарной исходному образцу.
Секвенирование проходит с помощью лигирования восьми-нуклеотидных зондов, меченных на 5’-конце одним из четырех различных флуорофоров. Последовательность зондов несёт сайт гидролиза, находящийся между пятым и шестым нуклеотидами. Первые два основания (считая с 3’-конца) комплементарны двум нуклеотидам секвенируемой последовательности.
С третьего по пятый основания зонда могут гибридизоваться с любыми тремя нуклеотидами секвенируемой ДНК. 6-8 основания зонда также могут гибридизоваться с любой последовательностью, однако они вместе с флуоресцентным красителем отщепляются от зонда в ходе реакции.
Главным преимуществом этого метода является то, что каждый нуклеодит прочитывается дважды, что сильно повышает точность секвенирования.
Метод пиросеквенирования
Технология была разработана Полом Ниреном и его студентом Мустафой Ронаги в 1996 году. Она основывается на синтезе новой ДНК-цепи на иммобилизированной анализируемой матрице с участием полимеразы. В качестве затравки выступает праймер. Затем происходит поочерёдное добавление каждого типа дезоксинуклеотидтрифосфатов (A, G, C, T).
При встраивании нуклеотида стехиометрически высвобождается побочный продукт — пирофосфат (PPi). Он, в свою очередь, с участием фермента ATP-сульфурилазы в присутствии аденозин-5'-фосфосульфата (dATPαS) превращается в ATP. ATP — источник энергии для фермента люциферазы, превращающей люциферин в оксилюциферин с выделением света. Компьютерная программа осуществляет детекцию и анализ света.
Невовлечённые в синтез нуклеотиды и ATP под воздействием фермента апиразы распадаются, после чего запускается следующий цикл реакции, с новым нуклеотидом.
Ионное полупроводниковое секвенирование
Микролунки, содержащие предназначенную для секвенирования молекулу матричной ДНК, наполняют дезоксирибонуклеотидтрифосфатом (dNTP) одного вида. Если введенный dNTP является комплементарным к ведущему нуклеотиду матрицы, он включается в растущую комплементарную цепь.
Это вызывает высвобождение ионов водорода, который вызывает срабатывание ионного датчика, который указывает, что реакция произошла. Если в последовательности матричной цепи присутствует повтор одного нуклеотида, несколько молекул dNTP будут присоединены в одном цикле. Это приводит к увеличению количества образовавшихся ионов водорода и пропорционально более высокому электрическому сигналу.
Эта технология отличается от других технологий секвенирования тем, что не использует модифицированные нуклеотиды и оптические датчики. Ion semiconductor sequencing может также упоминаться как Ion torrent sequencing, рН-опосредованное секвенирование, или полупроводниковое секвенирование.
Технология ионного полупроводникового секвенирования позволяет исследовать большие объёмы данных, например: транскриптомы отдельных клеток и тканей, метагеномы микробных сообществ, малые РНК и проводить ChIP-seq.
Одномолекулярное секвенирование
Данный метод разработан компанией Pacific BioScinces. Идея метода состоит в определении последовательности ДНК за счёт наблюдения за работой единичной молекулы ДНК-полимеразы в реальном времени.
При этом ДНК-полимераза достраивает вторую цепь исследуемой молекулы ДНК, используя нуклеотиды, меченные различными флуорофорами; регистрируя данные метки, можно понять, какой нуклеотид ДНК-полимераза встраивает в настоящий момент.
Этот метод секвенирования позволяет получать очень длинные прочтения порядка десятков тысяч нуклеотидов. Кроме того, метод работает без предварительной амплификации образца посредством ПЦР. Одномолекулярное секвенирование работает с минимальным количество исходной ДНК.
Бисульфитный метод
Это название группы методов, в основе которых один принцип: определение паттерна метилирования ДНК посредством её бисульфитной обработки.
Под воздействием бисульфита остатки цитозина превращаются в остатки урацила, но при этом не затрагиваются метилированные остатки 5-метилцитозина. Далее изменённая последовательность сравнивается с исходной, цитозины и тимины разделяются в изученных последовательностях, и можно установить, какие CpG-динуклеотиды были метилированы.
Процесс, используемый для секвенирования ДНК, известен как метод обрыва цепи или метод Сэнгера. Он основан на модифицированной форме полимеразной цепной реакции.
Метод обрыва цепи (секвенирование по Сэнгеру)
Наряду с нуклеотидами и полимеразой, используемой в стандартном процессе ПЦР, подготовленная для реакции обрыва цепи среда содержит кроме четырех обычных нуклеотидов ДНК также и варианты каждого из них, так называемые дидезоксинуклеотиды.
Эти дидезоксинуклеотиды похожи на правильные нуклеотиды ДНК, но не содержат 3'-гидроксильной группы.
После того как дидезоксинуклеотид присоединен к растущей цепочке ДНК, ДНК-полимераза не может добавлять никаких других нуклеотидов. Таким образом, процесс репликации прекращается и полученный фрагмент ДНК прерывается.
Репликационная среда содержит только небольшое количество дидезоксинуклеотидных вариантов каждого из четырех нуклеотидов ДНК.
Во время прохождения полимеразной цепной реакции существует высокая вероятность того, что фермент полимеразы добавит немодифицированный нуклеотид к растущей цепи и что процесс репликации будет продолжаться. Но иногда полимераза связывает дидезоксинуклеотид с цепью и реакция прекращается.
В суспензии, содержащей миллиарды фрагментов ДНК, конечным результатом является серия фрагментов, заканчивающихся одним из четырех дидезоксинуклеотидов.
Все вместе эти фрагменты представляют собой все возможные расположения нуклеотидов A, C, G и T на синтезируемой нити.
Каждый из четырех вариантов дидезоксинуклеотида возможно пометить разными маркерами (например, красителем, который испускает свет с определенной длиной волны в ультрафиолетовом свете) для того, чтобы сделать различные нуклеотиды легко идентифицируемыми.
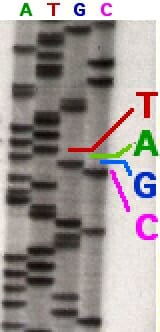
Поскольку фрагменты разделяются по длине и массе во время гель-электрофореза, маркеры указывают каким нуклеотидом заканчивается каждый фрагмент. Затем гель можно считывать снизу вверх для идентификации нуклеотидной последовательности.
Этот этап обычно включает в себя использование автоматического секвенатора ДНК, который ускоряет процесс считывания.
Метод секвенирования по Сэнгеру может быть использован для того, чтобы определить за одну реакцию последовательность ДНК образца, содержащего до одной тысячи пар оснований.
Этапы секвенирования больших геномов
Секвенирование большого генома включает в себя следующие три основных этапа:
1. Картирование генома.
Весь геном сначала случайно разбивается на более мелкие последовательности ДНК - приблизительно от 100 000 до 300 000 пар оснований.
Эти участки затем клонируют в бактериальном векторе, называемом бактериальной искусственной хромосомой или ВАС.
Повторяя этот цикл несколько раз, исследователи получают серию перекрывающихся ВАС.
Затем эти ВАС пропускают через гель-электрофорез, чтобы определить их индивидуальные фингерпринты ДНК. Изучив структуру этих паттернов, исследователи могут определить исходный порядок ВАС в геноме.
2. Секвенирование ДНК.
После того как исходный порядок ВАС был картирован, каждый ВАС разбивается рестрикционными эндонуклеазами на гораздо более мелкие фрагменты, которые могут быть секвенированы с использованием реакции обрыва цепи.
Этот этап секвенирования иногда называют BAC-BAC секвенирование.
3. Анализ результатов.
Паттерн перекрывающихся последовательностей ДНК используется для определения порядка фрагментов в каждом ВАС.
В этой процедуре используется несколько различных компьютерных программ, которые могут анализировать последовательности ДНК.
Секвенирование целого генома (метод дробовика, шотган-секвенирование, shotgun sequencing)

Этот метод полностью пропускает стадию картирования генома.
Вместо этого весь геном разбивается на случайные фрагменты сначала около 2000, а затем около 1000 пар оснований.
(Наличие фрагментов различной длины помогает сделать сборку нуклеотидных последовательностей более точной.)
Эти фрагменты, которые насчитывают миллионы, затем секвенируются и анализируются, после чего определяются нуклеотидные последовательности, соответствующие каждой из хромосомам.
Все это делается с помощью мощных компьютеров и сложных программ.
Секвенирование следующего поколения (высокопроизводительное секвенирование) (Next-generation sequencing, high-throughput sequencing)
Секвенирование следующего поколения (NGS) - это термин, используемый для описания ряда различных современных технологий секвенирования которые включают:
- Illumina (Solexa) sequencing
- Roche 454 sequencing
- Ion torrent: Proton / PGM sequencing
- SOLiD sequencing

Платформы NGS выполняют массовое параллельное секвенирование, в ходе которого миллионы фрагментов ДНК из одного образца секвенируются одновременно.
Технология массового параллельного секвенирования обеспечивает высокопроизводительное секвенирование, что позволяет секвенировать весь геном менее чем за один день.
В последнее десятилетие были разработаны несколько платформ NGS, которые обеспечивают недорогое и высокопроизводительное секвенирование.
Проект генома человека
Полный сиквенс человеческого генома был впервые опубликован в феврале 2001 года. Этот сиквенс был первым геномом млекопитающих, у которого была определена последовательность нуклеотидов в каждой хромосоме.
В результате проекта генома человека (HGP) была определена последовательность трех миллиардов пар оснований, которые составляют геном человека.
Среди непосредственных результатов проекта было открытие, что ДНК всех людей (Homo sapiens) более чем на 99,9% идентичны.
Другими словами, это означает, что все различия между людьми во всем мире являются результатом изменений менее чем одного нуклеотида на каждую 1000 в геноме каждого человека.
Методом Сэнгера секвенируют интересующий участок ДНК, когда известны точные координаты предполагаемого изменения в геноме после проведения полноэкзомного секвенирования.
Из любого города России
Результат на почту в течение 30 рабочих дней
Все данные и результаты не могут быть переданы третьим лицам
Секвенирование
Молекула ДНК является носителем генетической информации практически во всех живых организмах. Она состоит из двух комплементарных, то есть соответствующих друг другу цепей нуклеотидов, формирующих уникальную последовательность.
Пару комплементарных нуклеотидов в составе цепи ДНК также называют парой оснований, сокращенно п.о.
Человеческий геном содержит около 3-х миллиардов пар оснований.
Секвенирование — метод, который используется в лабораторной практике для определения последовательности ДНК.
В диагностике его часто используют для поиска изменений в структуре генов или их регуляторных элементов, вызывающих заболевания (патогенные мутации, варианты).
Секвенирование по Сенгеру
Показания
- для подтверждения данных, полученных методом NGS при выявлении патогенных вариантов или вариантов с неизвестной клинической значимостью.
- для установления генетического статуса пациента в случае, когда у кого-либо из родственников уже была выявлена мутация и ее точное положение в геноме известно.
Из-за большого объема анализируемых последовательностей при использовании метода NGS некоторые участки могут быть прочтены неточно, поэтому в случае обнаружения находок методом NGS рекомендуется подтверждать их с помощью секвенирования по Сэнгеру.
Особенности метода
В пробирке проводят реакцию синтеза ДНК по уже существующей матрице (одноцепочечной генетической последовательности конкретного индивида), последовательность которой нужно установить.
Молекулу ДНК синтезирует фермент ДНК-полимераза, которая, в соответствии с матрицей, по одному встраивает нуклеотиды в растущую цепь.
В результате образуется смесь уникальных фрагментов ДНК разной длины, которые начинаются одинаково, но заканчиваются в разных положениях вдоль цепи ДНК-матрицы. Причём заканчиваются они одной из четырёх нуклеотидных меток.
Затем происходит разделение этих фрагментов по величине с помощью капиллярного электрофореза.
Синтезированные фрагменты ДНК разделяются под действием электрического поля: чем меньше фрагмент, тем он быстрее движется в геле.
После происходит детекция флуоресцентного нуклеотида на конце каждого фрагмента и определяется полная нуклеотидная последовательность исследуемого образца ДНК.
Сравнение методов
С момента изобретения секвенирования по Сэнгеру продолжались поиск и разработка альтернативных способов расшифровки ДНК. В последнее время большой популярностью пользуются методы секвенирования нового поколения (NGS). Однако, у каждой технологии есть свои особенности и выбор метода обычно производится исходя из поставленной задачи.
Метод секвенирования по Сэнгеру имеет ограничения
С его помощью можно прочитать только последовательности ДНК до 1000 пар оснований, полученные от одного человека, за один раз.
Кроме того, невозможно детектировать изменения в некоторой популяции клеток, когда в большей части образца представлена нормальная копия гена. Например, в опухолевых клетках происходят изменения, вызывающие их бесконтрольный рост. Такие изменения не будут присутствовать ни в каких других клетках организма.
Секвенирование по Сэнгеру позволяет детектировать изменения только если они проявляются не менее чем в 15-20% исследуемой ДНК.
15-20% исследуемого материала должно содержать изменения, чтобы их можно было детектировать методом Сенгера
Методы секвенирования нового поколения
Позволяют получить данные о более длинных последовательностях ДНК (вплоть до полногеномного секвенирования) и детектировать варианты (мутации), содержащиеся примерно в 5% исследуемой ДНК. Однако это происходит за счет одновременного прочтения большого числа коротких (в среднем, для методов, наиболее часто используемых в клинических лабораториях, 100-300 п.о.) последовательностей ДНК, которые затем соединяются и анализируются.
Процесс обработки таких данных достаточно трудоемкий и требует высокого уровня квалификации специалиста.
В тех случаях, когда есть подозрения, что выявленный вариант является причиной заболевания, необходима 100% уверенность в правильном прочтении. Тогда имеет место подтверждение наличия патогенного или условно-патогенного варианта методом Сэнгера.
Также методы NGS являются дорогостоящими, из-за чего анализ с их помощью коротких последовательностей экономически невыгоден.
Поэтому, когда известны точные координаты предполагаемого изменения в геноме (например, если у кого-то из близких родственников был обнаружен патогенный или условно-патогенный вариант), проводить для него полноэкзомное секвенирование не обязательно. Можно просто секвенировать интересующий участок ДНК методом Сэнгера.
5% исследуемого материала должно содержать изменения, чтобы их можно было детектировать методом нового поколения (NGS)
Читайте также: