
Полимеризация нитроанилина как сделать
Добавил пользователь Skiper Обновлено: 19.09.2024
В видео показан классический метод синтеза п-нитроанилина из ацетанилида Все добавленные видео защищены .
Определение понятий и классификация реакций полимеризации и поликонденсации с примерами реакций. Канал "День .
Подробный разбор катионного механизма реакции полимеризации на примере изобутилена. 1) Полимеризация этилена .
имия, химические опыты, химические реакции, неорганическая химия, егэ по химии, органическая химия, химия просто, .
Check out the Most MINDBLOWING Chemical Reactions You Probably Haven't Seen Before! From surprising experiments .
Hello friends, in this video we synthesise p-nitroaniline from acetanilide via a two step reaction Preparation of Acetanilide .
В видео показан один из способов получения одного из изомерных нитротолуолов Все добавленные видео защищены .
In this video, I will be synthesizing p-nitroaniline from acetanilide. This chemical is used mainly as a precursor for a dye, but it can .
Уролог, андролог, врач УЗД, Доктор медицинских наук Филиппов Сергей Викторович отвечает на вопросы из ежемесячной .
В этом видео разберем различные варианты энергосбережения, как входить в режим энергосбережения и как из него .
Всем привет, представляю вашему вниманию очень полезный инструмент для трейдинга, я уверен ему найдется место в .



Нитросоединения жирного ряда могут быть получены при непосредственном действии азотной кислоты или окислов азота на предельные углеводороды. Прямое нитрование углеводородов жирного ряда и нафтеновых углеводородов изучено М. И. Коноваловым (реакция Коновалова). С. С. Наметкин объяснил механизм этой реакции и широко использовал ее для установления строения терпеновых углеводородов. Однако метод прямого нитрования мало пригоден для препаративных целей, так как он не дает возможности получить достаточно однородный продукт. Лучшие результаты получаются при действии азотистокислых солей на галоидопроизводные углеводородов, например:
В качестве побочного продукта при этом образуется некоторое количество сложного эфира азотистой кислоты C 2 H 5 -O-NO, который легко отделить от нитросоединения ввиду значительно более низкой температуры его кипения.
Нитросоединения ароматического ряда, напротив, легко получаются при непосредственном нитровании углеводородов и других ароматических соединений - фенолов, кислот и пр. В случае легко нитрующихся соединений реакция идет при применении разбавленной азотной кислоты; трудно вступающие в реакцию вещества нитруют смесью концентрированных азотной и серной кислот. Серная кислота связывает образующуюся при реакции воду и тем самым поддерживает необходимую для реакции концентрацию азотной кислоты. Непосредственное нитрование является единственным практически применяемым методом получения нитросоединений ароматического ряда.
Примером получения нитросоединений жирного ряда действием азотистокислых солей на галоидные производные может служить синтез нитрометана. В качестве исходного вещества берут монохлоруксусную кислоту, в которой хлор обладает значительной подвижностью. При взаимодействии натриевой соли хлоруксусной кислоты с азотнокислым натрием образуется натриевая соль нитроуксусной кислоты:
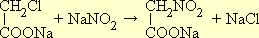
При нагревании нитроуксусная кислота легко декарбоксилируется (т. е. отщепляет CO 2 ), образуя нитрометан:
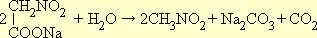
Реактивы:
Хлоруксусная кислота. 18,9 г (0,2 моля)
Азотистокислый натрий. 14 г (0,2 моля)
Едкий натр; хлористый кальций
К смеси указанного количества хлоруксусной кислоты и 20 г толченого льда прибавляют при помешивании 14 мл охлажденного во льду 40%-ного раствора едкого натра до щелочной реакции (по фенолфталеину). При нейтрализации температура смеси не должна подниматься выше 20°. Полученный раствор хлоруксуснокислого натрия приливают к раствору азотистокислого натрия в 18 мл воды, находящемся в перегонной колбе емкостью 200 мл. Колбу соединяют с холодильником и закрывают пробкой с термометром, шарик которого должен быть погружен в жидкость.
Смесь медленно нагревают до начала выделения пузырьков углекислого газа, что происходит при температуре около 80°. После этого прекращают нагревание, так как реакция продолжается самостоятельно, без подогрева извне. Если температура смеси начинает понижаться, то вновь осторожно подогревают до 85°. Когда реакция в основном закончится, реакционную смесь осторожно подогревают, доводя температуру ее к концу реакции до 110°. Нитрометан начинает перегоняться при температуре около 90°. При отгонке получают не менее 5 мл нитрометана и около 15 мл воды.
Темп. кип. чистого нитрометана 101,2°; уд. вес 1,1382; показатель преломления 1,3935.
Нитрометан является нейтральным соединением, но в присутствии щелочей изомеризуется в аци-форму, взаимодействующую со щелочью с образованием соли:
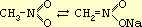
При подкислении раствора соль разлагается и выделившаяся в свободном виде аци-форма постепенно переходит в нейтральный нитрометан. Присутствие аци-формы нитрометана может быть обнаружено по характерному окрашиванию с хлорным железом.
Растворяют 0,5 мл нитрометана при нагревании в 1 мл 1 н. раствора едкого натра и прибавляют несколько капель раствора хлорного железа. По охлаждении в пробирку осторожно по каплям прибавляют разбавленный раствор соляной кислоты. Появляется кровавокрасное окрашивание.
Обязательным условием успешного протекания реакции является хорошее перемешивание реагирующих веществ.
Наряду с нитробензолом образуется небольшое количество динитробензола. Повышение температуры во время нитрования приводит к образованию значительных количеств динитропродукта.
Реактивы:
Бензол. 18 мл или 15,6 г (0,2 моля)
Азотная кислота уд. веса 1,4. 20 мл (0,28 моля)
Серная кислота конц. 25 мл (0,45 моля)
Углекислый натрий; хлористый кальций
В колбе емкостью 260 мл осторожно, при охлаждении, смешивают азотную кислоту с серной. К охлажденной до комнатной температуры смеси постепенно, небольшими порциями, прибавляют бензол, каждый раз хорошо перемешивая содержимое колбы и наблюдая за тем, чтобы температура смеси не превышала 50-60°. В случае необходимости колбу охлаждают водой. (Для уменьшения потерь бензола за счет испарения к колбе присоединяют воздушный холодильник.) Когда весь бензол прибавлен, колбу помещают на водяную баню, нагретую до 60°, и ведут реакцию при этой температуре в течение получаса, часто и энергично перемешивая жидкость.
Затем реакционную смесь переливают в литровую колбу, содержащую 300 мл воды, перемешивают жидкость, охлаждают и при помощи делительной воронки отделяют находящийся в нижнем слое нитробензол *1 . Его промывают в делительной воронке сначала разбавленным раствором углекислого натрия, а затем чистой водой.
Промытый нитробензол переливают в небольшую колбу и прибавляют прокаленный хлористый кальций. Колбу закрывают пробкой, в которую вставлена стеклянная трубка (в качестве обратного воздушного холодильника), и нагревают на водяной бане. Когда жидкость станет прозрачной, переливают нитробензол в перегонную колбу и перегоняют с воздушным холодильником. После отгонки небольшого количества непрореагировавшего бензола перегоняется нитробензол при температуре 204-207°. Отгонять продукт досуха не следует во избежание разложения остающегося в колбе динитробензола *2 .
Темп. кип. чистого нитробензола 210,9°; уд. вес 1,2055; показатель преломления 1,5532.
Согласно правилу замещения в бензольном ядре, вторая нитрогруппа вступает в мета-положение по отношению к первой:
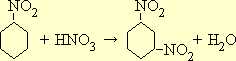
Так как замещение второго водородного атома бензольного ядра на нитрогруппу идет значительно труднее, чем замещение первого, то при получении динитробензола приходится создавать более жесткие условия нитрования (повышенная концентрация кислот, более высокая температура).
При реакции, кроме основного продукта м -динитробензола, получаются небольшие количества п -динитробензола (около 3%) и о -динитробензола (около 1%), которые можно удалить путем перекристаллизации препарата из спирта.
Реактивы:
Нитробензол. 12,3 г (0,1 моля)
Азотная кислота уд. веса 1,4. 10 мл (0,14 моля)
Серная кислота конц. 30 мл (0,55 моля)
Углекислый натрий; спирт
В колбу емкостью 200 мл вливают нитробензол и нагревают под тягой на кипящей водяной бане. К горячему нитробензолу при энергичном перемешивании небольшими порциями прибавляют нитрующую смесь. Температура реакционной смеси во время нитрования не должна подниматься выше 115°.
После внесения всего количества нитрующей смеси нагревание и перемешивание продолжают еще в течение 30-40 мин. Конец реакции устанавливают на основании следующей пробы: каплю раствора вносят в пробирку с водой; динитробензол должен при этом выпадать в виде бледножелтых кристаллов; если этого не происходит, нагревание необходимо продолжить.
По окончании реакции смесь охлаждают до 70° и при энергичном перемешивании ( под тягой ) выливают в 100 мл холодной воды. Сырой динитробензол выпадает в виде аморфной массы. По охлаждении кислый раствор декантацией сливают с осадка *1 , добавляют к последнему 50 мл воды и нагревают до кипения; динитробензол при этом плавится. По охлаждении воду сливают и повторяют ту же операцию, добавляя к воде углекислый натрий до резко щелочной реакции (по лакмусу). Охладив раствор, сливают воду через фильтр, а оставшийся на дне стакана динитробензол (в виде твердой лепешки) еще два раза плавят в чистой воде, беря каждый раз по 50 мл воды и сливая охлажденный раствор через тот же фильтр. Небольшое количество задержанных фильтром кристаллов промывают холодной водой, отжимают между листами фильтровальной бумаги, присоединяют к основной массе динитробензола, который вынимают из стакана, и высушивают на воздухе.
Полученный продукт плавится около 80°. Для получения вполне чистого м -динитробензола его перекристаллизовывают из спирта. Он образует бесцветные длинные иглы с темп. пл. 90°.
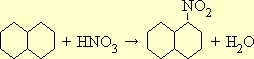
Реактивы:
Нафталин. 12,8 г (0,1 моля)
Азотная кислота уд. веса 1,4. 7,2 (0,1 моля)
Серная кислота конц. 13 мл (0,23 моля)
Метиловый спирт
Серную кислоту смешивают с 7 мл воды и с указанным количеством азотной кислоты. К смеси, нагретой до 50°, прибавляют тонко растертый нафталин и, поддерживая указанную температуру, ведут реакцию в течение 1 часа при постоянном перемешивании. (Целесообразно применить механическую мешалку.) Затем повышают температуру до 60° и в течение часа продолжают перемешивание смеси.
По охлаждении нитронафталин застывает в виде лепешки, плавающей на поверхности раствора. Кислую жидкость сливают, а сырой нитронафталин плавят в кипящей воде. По охлаждении воду сливают и повторяют эту операцию еще два раза. При такой обработке большая часть непрореагировавшего нафталина улетучивается с парами воды. Расплавленный продукт при энергичном перемешивании выливают в холодную воду, в которой он застывает в виде маленьких шариков. Осадок отфильтровывают, отжимают между листами фильтровальной бумаги и сушат на воздухе.
Полученный продукт не вполне чист и содержит небольшие количества динитронафталина и непрореагировавшего нафталина. Для получения чистого препарата его кристаллизуют из метилового спирта: он выпадает в виде желтых игл с темп. пл. 61,5°.
Фенол нитруется очень легко уже на холоду под действием разбавленной азотной кислоты. В соответствии с ориентирующим влиянием OH-группы как заместителя 1-го рода при этом получается о - и п -нитрофенол:
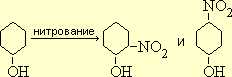
Разделение этих изомеров основано на том, что о -нитрофенол, в отличии от п -нитрофенола, перегоняется с водяным паром.
При нитровании фенола нужно избегать повышения температуры реакционной смеси во избежание образования ди- и тринитрофенола.
Реактивы:
Фенол. 28,2 г (0,3 моля)
Азотная кислота. 185 мл (0,6 моля)
Метиловый спирт; едкий натр; активный уголь; соляная кислота
К фенолу прибавляют 5 мл воды, нагревают до плавления, и смесь постепенно при перемешивании вносят в азотную кислоту. Колбу с азотной кислотой охлаждают водой, наблюдая за тем, чтобы температура реакционной смеси все время была ниже 20°. Смесь, принимающую темную окраску, оставляют стоять в течение нескольких часов в холодной воде, периодически взбалтывая.
По окончании реакции тщательно сливают кислоту, промывают несколько раз водой оставшуюся в колбе маслянистую, частично осмолившуюся массу и подвергают ее перегонке с водяным паром (прибор собирают, как показано на рис. 18). В приемник в виде желтого быстро кристаллизующегося масла переходит о -нитрофенол. Если о -нитрофенол начинает кристаллизоваться в холодильнике, то на некоторое время прекращают подачу в него воды; горячий конденсат расплавляет кристаллы о -нитрофенола, и он переходит в приемник.
Выпавший в приемнике о -нитрофенол отфильтровывают на воронке Бюхнера, отжимают между листами фильтровальной бумаги и высушивают на воздухе.
Если полученный продукт плавится при более низкой температуре, его перекристаллизовывают из метилового спирта.
Для выделения п -нитрофенола оставшуюся в колбе смолистую массу кипятят с 170 мл 10%-ного раствора едкого натра и небольшим количеством активного угля и фильтруют.
Еще горячий темный фильтрат упаривают до тех пор, пока капля раствора по охлаждении не будет застывать. Раствор охлаждают, выделившийся п -нитрофенолят отсасывают, промывают несколько раз небольшими порциями 10%-ного раствора едкого натра и хорошо отжимают на фильтре.
Полученную соль переносят в стакан и при нагревании разлагают 10%-ной соляной кислотой. Выделившийся нитрофенол по охлаждении застывает. Водный слой сливают и перекристаллизовывают нитрофенол из горячей 1-2%-ной соляной кислоты.
Свободный анилин при нитровании легко подвергается окислению и осмолению. Для предохранения аминогруппы от окисления анилин сначала подвергают ацетилированию. Полученный ацетанилид нитруется с образованием преимущественно п -нитроацетанилида:
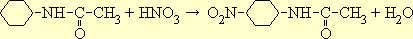
Если нитрование ведется при низкой температуре, то изомерный о -нитроацетанилид образуется лишь в весьма небольших количествах; повышенная же температура благоприятствует его образованию.
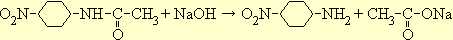
Реактивы:
Ацетанилид. 13,5 г (0,1 моля)
Азотная кислота уд. веса 1,38. 8 мл (0,11 моля)
Серная кислота конц.; углекислый натрий; спирт; едкий натр
Тонко измельченный сухой ацетанилид вносят в 30 мл концентрированной серной кислоты и перемешивают до тех пор, пока не получится вполне прозрачный раствор. Температура при этом не должна подниматься выше 25° во избежание омыления ацетанилида.
Раствор охлаждают до 0° в смеси льда и соли, и постепенно приливают смесь 8 мл азотной кислоты и 5 мл концентрированной серной кислоты. Температура во время нитрования не должна превышать 2-3° во избежание образования значительных количеств о -нитросоединения. После того как прибавлена вся кислота, продолжают перемешивание еще в течение получаса и смесь оставляют стоять на холоду в течение ночи.
На следующий день вливают раствор в смесь 35 мл воды и 35 г толченого льда; тотчас выпадает нитроацетанилид. Через полчаса осадок отфильтровывают, хорошо промывают водой, переносят в стакан с 50 мл воды, прибавляют углекислый натрий до щелочной реакции (по лакмусу) и нагревают до кипения. При этой обработке о -нитроацетанилид омыляется, а п -нитроацетанилид остается без изменения. Охлаждают раствор до 50°, отфильтровывают кристаллы п -нитроацетанилида, хорошо промывают водой и высушивают на воздухе.
Для получения п -нитроанилина сырой п -нитроацетанилид смешивают с 20 мл воды, приливают 12 мл 35%-ного раствора едкого натра и кипятят до тех пор, пока капля раствора, внесенная в 10%-ную соляную кислоту, не будет растворяться без осадка.
Обычно омыление заканчивается через 2,5-3 часа. Нужно следить, чтобы во время кипячения реакция все время оставалась щелочной. Смеси дают охладиться до 40°, отфильтровывают осадок, тщательно промывают холодной водой и высушивают.
*1 Остающийся в делительной воронке водный слой, содержащий серную и азотную кислоты (а также аналогичные ему растворы), нельзя непосредственно выливать в канализационную раковину; предварительно его необходимо нейтрализовать известковым молоком или известью.

основания Шиффа с ароматическими альдегидами , дает карбиламинную пробу .
качественн ым реакциям . В случае анилина взаимодействие с азотистой
кислотой приводит к образованию относительно устойчивого фенилдиазония ,

2. Реакции по бензольному ядру для анилина , N- алкил ( диалкил ) анилинов
сильно активирующим бензольное яд ро и направляют электрофил в орто - и
происходит протонирование атомов азота этих групп , и они превращаются в
Группа -NHC(O)R относится к донорам средней силы , неподеленная пара
электронов атома азота находится в сопряжении с ароматическим ядром ,
сопряжение с карбонильной группой реализуется лишь частично , в отличие от
амидов алифатического ряда ( смотрите свойств а амидов ).

б ) Для получения моногалогенпроизводных анилина необходимо снизить
ацетильную группу снимают с помощью щелочного гидролиза .
Другим способом монобромирования является использование
диоксанбромида . Из - за большого объема реагента получают исключительно

а ) Прямое нитрование приводит к образованию смеси продуктов , части чному
осмолению реакционной массы и не имеет препаративного значения .
В начале из анилина и азотной кислоты образуется соль – нитрат анилиния .
яв ляется мета - ориентантом в следствии –I – эффекта . В
реакционной массе соль анилина находится в равновесии с исходным
анилином , поэтому кроме мета - нитроанилина получаются орто - и пара -
б ) Для того , чтобы получить орто - и пара - нитроанилины аминогруппу
защищают с помощью ацилирования . Далее нитруют нитрующей смесью или
конц . азотной кислотой в уксусном ангидриде . Затем ацетильную защиту

С помощью сульфирования получают промышленно важные аминобензол
сульфокислоты , которые используются в производстве красителей . Варианты
Алкилирование и ацилирование по Фриделю - Крафтсу анилина и его N-
алкил и N,N- диалкил производных по ароматическому ядру идут с трудом и не
являются препаративными си нтезами . Анилин в качеств е основания Льюиса ,
связывается с кислотами Льюиса , а в присутствии кислот протонируется , что

Соединения , содержащие диазогруппу N=N , называют азо или
В азосоединениях диазогруппа связана с двумя алкильными (R) или
Диазосоединения получают из аминов с помощью реакции
диазотирования . Реагент ами являют ся нитриты щелочных металлов в
Диазопроизводные ароматического ряда устойчивы до 65° С в отличие от
диазопроизводных алифатического ряда ( см . тему “ Амины ”). Причиной
является эффекти вная делокализация положительного заряда диазониевой
Исследования показали , что молекула фенилдиазония плоская
( диазогруппа линейно расположена в плоскости кольца ), связ ь межд у атомами
Анилин и другие ароматические амины легко окисляются под действием нитрующей смеси азотной и серной кислоты, а также раствора азотной кислоты в уксусной кислоте или уксусном ангидриде. Поэтому нитрованию подвергают ацильные производные аминов.
При проведении реакции нитрования можно изменять:
- Электрофильную силу нитрующего реагента;
- Концентрацию нитрующего реагента;
- Температурный режим и продолжительность процесса;
- Растворитель.
При этом наблюдаются интересные различия в ориентации для нитрующих агентов различной природы.
Этот пример отчетливо выявляет стремление к нитрованию в орто-положение в системе азотная кислота - уксусный ангидрид. Ацильную группу, как обычно, удаляют кислотным гидролизом.
Третичные амины, можно нитровать под действием азотной кислоты в уксусной кислоте.
Получение орто-нитроанилина
Метод получения орто-нитроанилина заключается в блокировании пара-положения с помощью сульфогруппы с последующим нитрованием по орто-положению. Гидролиз 2-нитроанилин-4-сульфокислоты действием 60%-ной водной серной кислоты дает орто-нитроанилин.
Готовые работы на аналогичную тему
Общие закономерности нитрования
Нитрогруппа - важная функциональная группа в химии ароматических соединений, которую можно превратить во много других функциональных групп, поэтому реакция нитрования является путем к различным замещенным ароматическим соединениям. Многие свойства нитрогруппы можно объяснить на основании резонансного гибрида двух структур Льюиса.
В этих структурах система $O - N - O$ подобна $\pi$-системе алильного аниона.
Общепринято, что активной электрофильной частицей в реакции нитрования является нитроний-катион, образующийся в нитрующей смеси по уравнению:
Криоскопическим методом было доказано, что в нитрующей смеси существует не три, а четыре частицы, экспериментально наблюдается четырехкратная депрессия точки замерзания. В смеси азотной и серной кислот устанавливается равновесие, в которой присутствуют большое количество частиц. Одной из них является нитроний-катион $^+$, который был зафиксирован спектроскопическими методами. В смеси кислот он образуется в процессе, в котором серная кислота играет роль кислоты, а азотная - основания.
Строение иона нитрония подобно изоэлектронной молекуле диоксида углерода. Эта молекула линейная и является сильным электрофильным реагентом.
Рисунок 7. Нитроний-катион
Ион нитрония реагирует непосредственно с бензольным ядром с образованием интермедиата - пентадиенильного катиона.
Реакция по атому кислорода дает нитриты $R-O-NO$. Нитриты являются неустойчивыми при таких сильнокислых условиях и разлагаются с образованием продуктов, содержащих связи карбон - кислород. Эти продукты окисления дальше реагируют с образованием глубоко окрашенных полимерных соединений В большинстве реакций ароматического нитрования образования большего или меньшего количества смолоподиб ных побочных продуктов является обычной побочной реакцией.
Агенты нитрования
Существование нитроний-катиона доказано спектрально, а некоторые из его солей удалось выделить:
Они также представляют собой очень сильные нитрующие агенты. Замена серной кислоты на $HClO_4$, $HF$ и $BF_3$ также дает эффективные нитрующие смеси. Незначительный эффект самой азотной кислоты объясняется незначительным содержанием нитроний-катиона, образующегося по реакции
К мягким нитрующим агентам относятся азотная кислота в смеси с уксусной кислотой, ацетил- и бензоил-нитратами можно нитрувать пятичленные гетероциклы. Очень мягким реагентом нитрования является тетранитрометан в пиридине.
Другой активный агент нитрования - ацетилнитрат - образуется при растворении азотной кислоты в уксусном ангидриде:
Реакция нитрования является общей как для активированных, так и не активированных субстратов. Введение одной нитрогруппы значительно снижает активность к дальнейшему замещению, поэтому часто можно получить продукт мононитрование, но динитрование также возможно.

К электропроводящим относятся полимеры, которые могут быть как полупроводниками, так и проводниками (как металлы). Проводящие полимеры совмещают в себе гибкость и прочность пластиков с электропроводящими свойствами, характерными для металлов, и обладают гигантским потенциалом для практического применения. Основным преимуществом электропроводящих полимеров является их технологичность, т.к. они являются пластмассами и, следовательно, могут сочетать такие механические свойства, как гибкость, прочность, ковкость, эластичность с высокой электропроводностью. Проводящие полимеры получают все более широкое использование в различных электронных и электрических устройствах и областях, таких как датчики, светоизлучающие диоды, антистатики, электромагнитное экранирование и т.д. Среди различных видов проводящих полимеров наиболее широко используется анилин благодаря своей высокой стабильности и технологичности. В настоящее время рассматриваются возможности использования полупроводящих полимеров в качестве полупроводниковых приборов: светодиодов, транзисторов, что позволит резко расширить сферы их использования.
Одним из наиболее перспективных электропроводящих полимеров, который интенсивно исследуется с момента открытия его полупроводниковых свойств, является полианилин (ПАНИ). Это связано с тем, что данный полимер может находиться в разных состояниях окисления и, также как неорганические полупроводники, дает закономерный отклик на внешнее воздействие. Полианилин меняет свою электропроводность, цвет, плотность, магнитные свойства, гидрофильность-гидрофобность, проницаемость для газов и жидкостей.
Полианилин обладает контролируемой электронной проводимостью в диапазоне 10 -10 −10 1 См/см в сочетании с ионной проводимостью, окислительно-восстановительной активностью, электро- и сольватохромизмом, нелинейными оптическими свойствами, парамагнетизмом. В дополнении к этому полимер не токсичен, устойчив в агрессивных химических средах, имеет высокую термическую стабильность и низкую себестоимость [1, 2].
Чтобы превратить полианилин в электропроводящий полимер его модифицируют химически или электрохимически, т. е. допируют. Полимеризацию анилина можно проводить электрохимическим путем и под воздействием ферментов и химических веществ: персульфата аммония, хлорида железа (III) и бихромата калия. Полианилин существует в трех формах: эмеральдин, пернигранилин и лейкоэмерельдин. Протонированный эмеральдин, полученный в результате полимеризации, может быть окислен до пернигранилина и восстановлен до лейкоэмеральдина. В зависимости от условий окисления и степени протонирования кислотами формы полианилина связанны между собой обратимыми переходами. Эти превращения сопровождаются изменением цвета.
При электрохимическом синтезе полианилина в эмеральдиновой форме часто получают в виде тонких пленок электрохимическим окислением анилина в водных кислых средах на металлических или стеклянных проводящих электродах. Значение разности потенциалов при полимеризации составляет -0,2 - 0,9 В [2].
Полианилин получают в результате окислительной полимеризации анилина под действием различных окисляющих агентов. Чаще всего полимеризацию анилина проводят в водных растворах с использованием таких инициаторов, как: персульфат аммония, бихромат калия или хлорид железа (III). Как правило, реакцию проводят в сильнокислой среде при рН от 0,0 до 2,0, используя стехиометрически равные концентрации мономера и окислителя, чтобы избежать деградации (переокисления) полимера.
На данный момент наибольшее распространение получил метод синтеза ПАНИ в результате окислительной полимеризации анилина под действием различных окисляющих агентов. Чаще всего полимеризацию анилина проводят в водных растворах с использованием таких инициаторов, как: персульфат аммония, бихромат калия или хлорид железа (III). Наибольшее распространение получил метод синтеза полианилина в результате полимеризации анилина в солянокислом водном растворе под действием персульфата аммония. В результате этой реакции выход полианилина составляет максимальные значения (около 90-95%), а его удельнаяэлектропроводность имеет довольно высокие значения (100-500 См/м) [2]. Основными преимуществами химического синтеза являются: его простота и возможность получать большие количества ПАНИ с высоким выходом, а также низкая стоимость окислителя [3]. Основным недостатком метода являются: получение нерастворимого в обычных растворителях порошка ПАНИ и вытекающая отсюда сложность формирования проводящих слоев на соответствующих носителях.
Для получения ПАНИ в результате химического синтеза (см. рисунок 1) персульфат калия растворяют в дистиллированной воде и перемешивают в течение 15 минут. Анилин добавляют по каплям при постоянном перемешивании. Далее проводят термостатирование при температуре 30 в течение 5–48 часов [4]. После осадок отфильтровывают через фильтр Шотта и промывают дистиллированной водой до нейтрального pH. Полученный осадок проходит термическую обработку при температуре 105 о С в течение 60 мин.
ПАНИ также может быть получен химическим окислением анилина без добавления какой-либо кислоты. Данный способ является альтернативным способом, позволяющим получать ПАНИ без использования специальных высококислотных реактивов, обладающих коррозионной стойкостью и аммиака для нейтрализации полимера, что уменьшает трудоемкость данного способа и сделает его более безопасным для окружающей среды.
Рисунок 1. Технологическая схема получения полианилина
химическим методом: 1 - реактор-смеситель с рубашкой; 2 - отстойник (осадитель); 3 - фильтр; 4 - декантатор; 5 - сушилка
Другим альтернативным способом получения ПАНИ является электрохимическая полимеризация, которая заключается в получении тонких пленок (реже порошка) полианилина электрохимическим окислением анилина в водных кислых средах на металлических или стеклянных проводящих электродах. Обычно для этого используют метод циклической вольтамперометрии, который позволяет исследовать процессы восстановления и окисления, протекающие на одном и том же электроде в одном растворе. Реже используется синтез проводящего слоя полианилина из раствора анилина, находящегося в контакте с сильной кислотой [4].
Электрохимический синтез ПАНИ позволяет получить более чистый продукт, свободный от содержания примесей окислителя, дает возможность контроля толщины наносимого слоя при оптимальном подборе условий с использованием различных физических методов анализа (например, спектроскопию, хронокулонометрию и т.д.) непосредственно в ходе процесса осаждения слоя [6, 7]. Также электрохимическая полимеризация анилина позволяет получить ПАНИ заданной степени окисления и характеризуется минимальным количеством или полным отсутствием побочных продуктов в зависимости от условий полимеризации. Недостатками данного метода являются: возможность осуществления полимеризации анилина только на поверхности электрода, ограничения по выбору материала подложки для конкретного синтеза и по площади рабочего электрода, а также необходимость проведения большого количества электрохимических циклов полимеризации для получения значительных количеств ПАНИ (например, нескольких десятков граммов), что существенно затрудняет и удорожает его синтез [8]. Кроме того, метод химического синтеза далек от экологически благоприятного, так как протекает в сильнокислой среде и требует больших количеств окислителя, продукты восстановления которого необходимо утилизировать.
В результате химического или электрохимического синтеза получают макромеолекулы ПАНИ, которые формируют систему полисопряжения в результате строгого чередования бензольных колец и атомов азота, находящихся в основной полимерной цепи. Носители заряда - положительные поляроны - вводятся в полимер путем его химического или электрохимического окисления [9].
В общем виде полианилин состоит из повторяющихся N-фенил-п-фенилендиаминных и хинондииминных блоков:
Однако считается, что при синтезе полианилина происходит образование его различных форм:
Различные формы полианилина сильно отличаются по физико-химическим свойствам:
- лейкоэмеральдин представляет собой бесцветное вещество, медленно окисляющееся на воздухе;
- эмеральдин представляет собой полупроводниковое вещество зеленого цвета с хорошей проводимостью;
- нигранилин представляет собой полупроводниковое вещество черно-синего цвета с хорошей проводимостью;
- пернигранилин и его соль - неустойчивые сине-лиловые соединения;
- эмеральдиновое основание - темно-фиолетовое вещество, которое при протонировании сильными кислотами дает соль зеленого цвета с проводимостью около 1 См/см и выше.
Особенностью ПАНИ, которая делает его перспективным материалом для различных микро- и наносенсоров, является изменение химических и оптических свойств при процессах протонирования (допирования) и депротонирования (дедопирования), которые происходят при его взаимодействии с компонентами-окислителями [10].
Делокализация носителей заряда и повышение электропроводности полианилина происходит в результате стабилизации поляронов сильными кислотами. В зависимости от состояния окисления и степени протонирования кислотами ПАНИ может существовать в различных формах, связанных обратимыми переходами:
Наиболее важной формой полианилина, обладающей электропроводностью, является протонированное основание эмеральдина [11]:
Полианилин отличается довольно высокой термостабильностью. Полианилин в эмеральдиновой форме устойчив при нагревании до 200°С, при этом не меняется его химический состав и отсутствует потеря массы. При нагревании от 200 до 300 °С масса полианилина уменьшается приблизительно на 10%, при этом процентное содержание атомов углерода, водорода и азота в составе остается постоянным [12, 13].
При достижении температуры 600 - 800°С начинается окисление полианилина кислородом воздуха. При окислении происходит потеря более 80% массы полианилина в результате испарения образующейся воды, а также образования продуктов распада. Дальнейшее нагревание до 1000°С вызывает незначительное уменьшение массы полимера до 93 - 95%.
При переходе от формы к форме ширина запрещенной зоны ПАНИ контролируемо меняется в диапазоне от 0,7 до 4.0 эВ [2]. Одновременно в широком диапазоне меняются электропроводность полимера его оптические, магнитные и поверхностные свойства (см. таблицу 1) [13].
Читайте также: